In tech, Moore’s law is the optimistic benchmark: computing performance improves rapidly over time (more capability for less cost). In drug R&D, the long-run trend has often looked like the mirror image. This is captured by Eroom’s law (“Moore’s law in reverse”), a shorthand for the observed decline in R&D efficiency. It is commonly measured as fewer new drugs approved per inflation-adjusted R&D dollar over decades, according to Scannell et al.
The point isn’t that past approaches were “wrong.” They were simply built for a different era. Traditional one-factor-at-a-time (OFAT) experimentation works when systems are low-dimensional and interactions are limited. But today’s R&D problems are increasingly multivariable and interaction-heavy—conditions where OFAT becomes an unsustainable trajectory, because it scales poorly and misses important factor interplay.
Now zoom in from the industry-level narrative to the day-to-day reality of an R&D lab. Software has evolved fast, but the lab workflow is still constrained by a mix of manual work and uneven hardware maturity. Some instruments are increasingly integration-friendly (APIs, modular setups, easier data handoffs), while others are legacy, closed systems that are hard to connect and even harder to adapt without custom engineering.
As a result, a lot of experimental work remains manual, repetitive, and error-prone. Many teams still operate in cycles of pipette → run → record → repeat, with decisions shaped by habit, intuition, or “what worked last time.” The issue isn’t that scientists are doing it wrong, but it’s that chemistry is inherently high-dimensional. Human bandwidth reaches saturation quickly under the dual pressure of mapping non-linear chemical interactions while simultaneously managing data that is fragmented across instrument files, notebooks, and spreadsheets. In this reality, the bottleneck becomes decision-making at scale: not lack of expertise, but lack of time, connectivity, and a system that can learn continuously from every run.
The transition toward a modern lab begins with lab digitalization. Not merely as a storage solution, but as a way to operationalize AI directly inside R&D workflows. Data is captured as it’s generated. Models update in real-time. The next experiment is recommended in context, not as an offline afterthought. And crucially, this shift doesn’t require chemistry teams to become full-time developers. The point is to lower the activation energy: chemists keep doing chemistry while AI tools handle the iterative optimization and decision support through accessible interfaces (often no-code), with optional API/SDK hooks when integration is needed.
- Beyond just speeding computation, AI is improving experimental decision-making.
- Better decisions mean fewer experiments, lower reagent burn, and lower operational cost.
- Closing the design–make–test loop is where value compounds.
- AI augments chemists. It does not replace scientific judgment, so chemistry teams will be the main characters of this story.
Digitization and automation of R&D workflows
In many lab settings, lab data is still trapped in silos: instrument exports here, ELN notes there, spreadsheets everywhere, and half the context living in someone’s head. While this fragmentation is a storage issue, it’s also an opportunity cost, or productivity tax: it slows learning, makes results harder to reproduce, and turns every new optimization into another round of manual guesswork, or time lost to actual discovery.
The fix starts with digitization that connects the workflow – consolidating experimental, analytical, and literature data into a shared, structured layer where context (conditions, metadata, outcomes) is captured consistently and remains reusable. This is the main idea behind making data Findable, Accessible, Interoperable, and Reusable (FAIR), because stored isn’t the same as usable.
Alongside reducing data silos, the real shift happens when experimental data begins to drive experimental decisions. In chemistry, the core challenge is rarely data storage – it is choosing the next experiment in a landscape shaped by many interacting variables such as catalysts, ligands, solvents, pressure, temperature, and time. As dimensionality increases, intuition has its limit, making it difficult to map every hidden interaction without a data-driven guide. What chemists need are tools that make advanced optimization and statistical learning part of the experimental workflow – without turning every project into a mental and software exercise. Sequential, model-guided experimentation has been shown to outperform manual experiment selection in both efficiency and consistency, particularly in complex, multivariable systems.
Crucially, this transition does not require laboratories to first “fix everything” upstream. In practice, digitization, automation, and AI adoption evolve in parallel. As context is captured more systematically (conditions, metadata, and outcomes), automation can be introduced where it adds value, and decision-making can shift from human-only heuristics toward data-informed iteration. Shields et al. state that this fact accelerates design–make–test cycles even in labs that are only partially automated, rather than forcing an all-or-nothing move toward full robotics.
Atinary’s AI platform SDLabs® operates within this reality as an enabling layer rather than a replacement workflow. It allows chemists to apply Bayesian optimization directly to experimental problems while keeping their attention on chemistry: defining constraints, interpreting outcomes, and steering discovery. The power of this shift is more evident and tangible whenever the search space becomes truly combinatorial – a challenge found across the entire R&D spectrum, from fundamental discovery to industrial scale-up. A prime example is hydroformylation optimization: here, an approach based on FalconGPBO explored a vast condition space and converged on optimal catalyst performance using just 88 physical experiments, illustrating how AI-guided experimentation can replace exhaustive trial-and-error with sample-efficient learning, delivering significant resource savings in a fraction of the traditional timeline
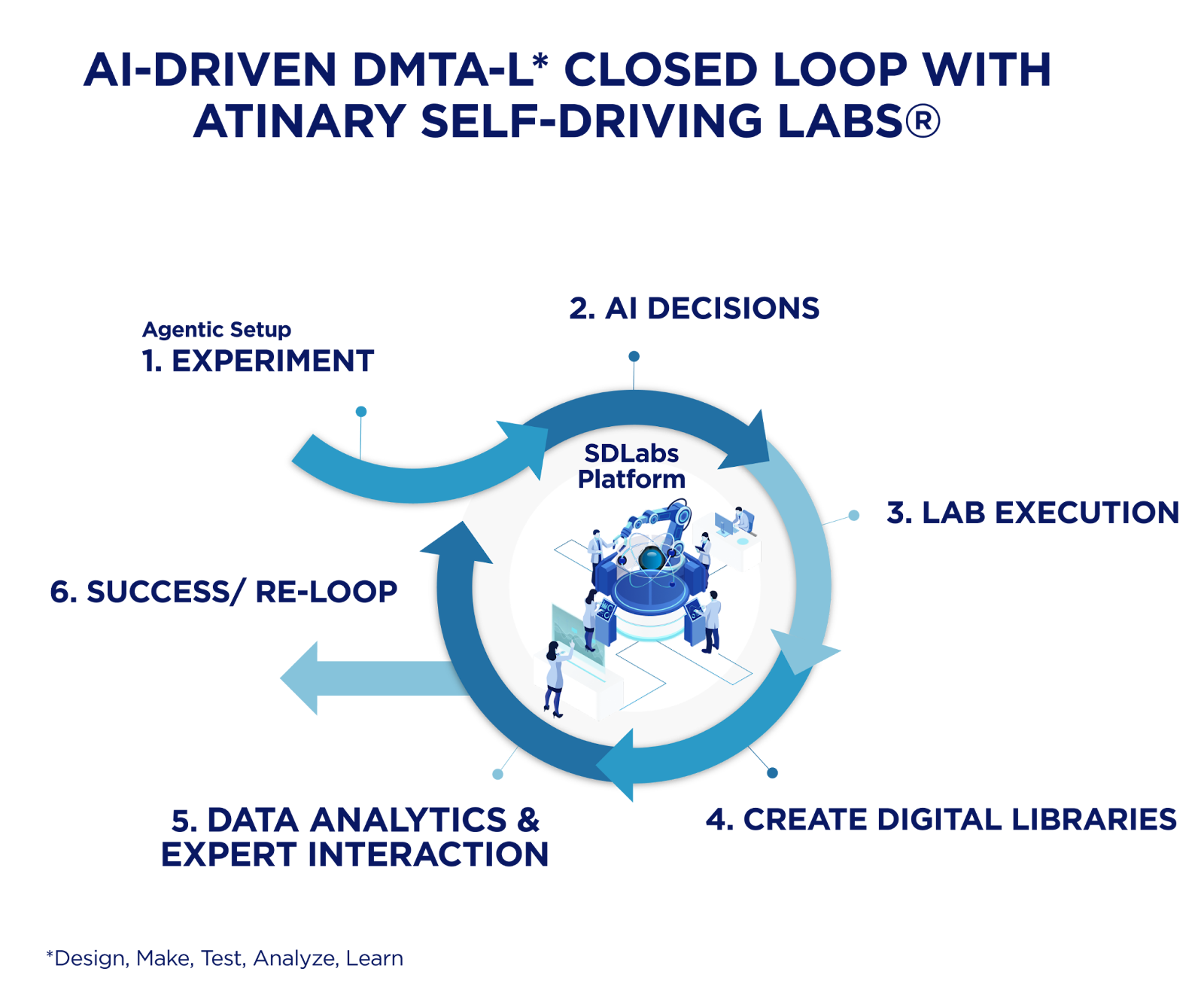
Closed-loop experimentation in self-driving laboratories
The foundation of this main shift is modularity. While the leap to a fully autonomous lab can be daunting, the path forward does not need to be an immediate, fully rigid loop, but a modular, hardware-agnostic integration that grows with your team’s readiness. As demonstrated by MacLeod et al., modular robotic platforms like “Ada” can autonomously optimize complex thin-film materials by iterating through diverse processing and compositional parameters in a fully autonomous loop. This approach allows R&D organizations to modernize iteratively, whether it is starting with a simple-to-use AI tool for designing the experimental space, or semi-automated workflows and scaling toward tighter loops as their data plumbing and analytical instruments are ready to be connected.
But a closed-loop system doesn’t have to be binary: you have it, or you don’t. This is further evidenced by Dai et al., who described a setup where a mobile robot interfaces with distributed, off-the-shelf lab equipment (e.g., synthesis hardware plus UPLC-MS and benchtop NMR) through standardized control software. Hence, by leveraging these flexible architectures, the workflow scales without the need for custom, monolithic “one lab, one robot” builds.
A compelling example of this transition is seen with Takeda’s HTE & Automation team, who demonstrate what “ahead of the curve” looks like in practice. By combining robotics, automated data handling with an AI layer (SDLabs®), they successfully removed traditional bottlenecks, compressing a 1-2 month manual optimization cycle down to about a week to optimize Buchwald-Hartwig reactions.
The success of such integrated workflows – as seen with Takeda – highlights a fundamental reality: the loop is only as powerful as the underlying data plumbing. To achieve this level of acceleration, analytical data must flow seamlessly, ensuring that inconsistent formats or fragmented silos do not stall the momentum before the chemistry gets interesting. The clean path forward is hardware-agnostic integration: connect instruments and analytics to the decision layer via API/SDK, so teams start with semi-automated workflows and scale toward tighter loops as readiness grows. For instance, the integration of Chemspeed automation and Bruker NMR demonstrated in Atinary’s demo lab setup serves as a practical blueprint for this, demonstrating how existing hardware can be unified into a high-performance, closed-loop system for applications in Suzuki and Buchwald-Hartwig reactions.
Looking ahead, it is worth noting that API/SDK-based connections could be increasingly augmented or partially replaced by Model Context Protocols (MCPs). This LLM-based orchestration enables different tools to exchange context and coordinate actions more fluidly, providing a flexible alternative to hard-wired, one-off integrations.
AI for exploring chemical design space
Building on the results mentioned earlier, the hydroformylation case study offers a definitive look at this efficiency in action. Here, BO guided a seven-dimensional reaction space of nearly 3 billion possible conditions, converging on optimal catalyst performance in just 88 experimental runs. This approach enabled a 10-30x reduction in rhodium catalyst loading, cut reaction time by 50% (from 16 to 8 hours), and reduced the rhodium cost contribution by >95%. This stands as a testament to how AI-driven optimization delivers resource-efficient outcomes under tight experimental constraints.
Beyond catalysis, these same optimization principles have been successfully applied to other complex process chemistry. For instance, Atinary collaborated with Snapdragon Chemistry to optimize oligonucleotide synthesis. This process involves repeated coupling, deprotection, and washing steps, each highly sensitive to reagent choice, timing, and specific process conditions. SDLabs® coordinated multi-variable optimization across the cyclic, multi-step synthesis cycle, improving both yield and lowering cost compared to the prior R&D process.
The results? Over a three-month engagement, the AI-assisted workflow increased product yield by up to 18% and reduced cost by approximately 22%. While these synthesis workflows are often performed using robotic platforms – such as those from Chemspeed or Unchained Labs – they can be further enhanced by the seamless integration and optimization provided by the SDLabs® API-friendly layer.
Autonomous synthesis and process optimization
In the Schilter et al. study, this approach showed how a closed loop can connect automated synthesis with in-line analysis and BO. By leveraging iterative, model-guided selection, the system converges quickly on a solution without exhaustively screening the entire design space. The primary payoff is operational: reaching target performance with only a small fraction of the potential experiments.
In the ETH Zurich’s SwissCat+ collaboration, SDLabs® guided an AI-driven workflow to identify an optimal catalyst formulation for CO₂-to-methanol conversion. The team was faced with 20M+ potential combinations across 11 parameters, 7 constraints, and 4 objectives . Rather than going through an endless screening phase, they ran the loop in a disciplined iterative manner: 144 combinations were tested across 6 iterations (24 batches per iteration), reaching compelling results and objectives by exploring just 0.00072% of the total combinatorial space.
What these results reveal is a paradigm shift: automation and algorithms handle the high-dimensional search, while chemists focus on strategic orchestration. In this model, the scientist’s role is elevated to setting the right objectives, enforcing complex chemical constraints, and providing the critical domain expertise to validate that the results align with physical reality. This is about moving from manual labor to high-value discovery, modernizing the lab by connecting existing automation into a unified, intelligent workflow. This evolution is explored extensively in our 2025 CHIMIA review on the promises and challenges of Self-Driving Labs as sustainable drivers for the future of chemistry.
And you don’t need to buy a fresh “robot empire” to get started. The Atinary demo lab with Chemspeed, for instance, showcases how organizations can get started by connecting the automation they already have. By integrating existing platforms like Chemspeed’s Flex iSynth) and analytics such as Bruker’s NMR into a closed-loop workflow using SDLabs®, teams can modernize iteratively, scaling their capabilities at a pace that matches their organizational readiness.
Safety, sustainability, and responsible use of AI
As Seifrid affirmed, by shifting repetitive, error-prone handling into standardized, automated execution, we reduce human exposure to sensitive and hazardous materials. This includes controlled environments such as gloveboxes or integrated workstations, where automation not only protects the researcher but also fundamentally improves reproducibility.
This operational safety can also be actively augmented by AI-driven monitoring systems. According to Munguia et al, platforms like Chemist Eye utilize vision-language models paired with RGB and infrared sensors to act as“digital guardians”. These systems provide situational awareness required for fully autonomous R&D by detecting incidents such as fire hazards or PPE non-compliance, sending real-time alerts, and even recommending corrective actions, allowing the lab to proactively move mobile robots away from risk areas and ensure the lab remains a safe space for both scientists and hardware.
Sustainability follows a similar logic to safety: both are built on the elimination or reduction of unnecessary risk and waste. When experimental optimization becomes sample-efficient, you are not just saving time, but also reducing solvent, catalyst, and consumables burned per learning cycle. In the former hydroformylation case, BO with Atinary Falcon reached resource-efficient conditions in 88 experiments, explicitly targeting reduced rhodium use and drastically lowering both costs and environmental footprint.
Beyond the bench, Responsible AI must also address security and operational readiness. In the enterprise world, adoption is often blocked by procurement and vendor-risk reviews rather than a lack of scientific curiosity. Enterprise-grade controls, auditability, and IP safeguards are table stakes, which is why benchmarks like SOC 2 compliance and availability via AWS Marketplace matter: they bridge the gap between cutting-edge technology and how regulated, IP-sensitive R&D teams actually buy and deploy software.
Data, trust, and regulation as challenges
This foundation of data quality leads to the second pillar: trust. In labs, a model that outputs a single best condition without confidence feels like a magic trick – especially when experiments are noisy and factor interactions are non-linear. Following this playbook, uncertainty quantification makes recommendations calibrated: what the model expects, how confident it is, and where it is essentially exploring new territory. As Zhou et al noted, interpretability matters too. Chemists don’t need to read the algorithm’s source code, but they do require transparent diagnostics, visual explanations, and sanity checks that map onto their chemical intuition.
Regulation is the third leg of the stool. In high-stakes R&D, trust also means governance: documentation, risk management, traceability, and clear human oversight of automated decisions. These expectations are increasingly formalized in risk-management frameworks like NIST’s AI RMF (NIST).
Hence, platforms must be structured with enterprise-grade security and operational controls, plus workflows that keep scientists in charge. Signals such as SDLabs® being SOC2 compliant and available via AWS Marketplace are aimed at that reality, ensuring that security, reliability, and IP sensitivity are “table stakes” for real-world deployment.
FAIR data principle impact on regulatory marks
Da Silveira et al. evaluated widely used chemical datasets. They found that even when raw data exists, incomplete metadata, non-standard formats, or fragmented records directly limit both: FAIRness and regulatory usability of the information. Therefore, aligning digitization efforts with FAIR principles does more than just prepare a lab for AI; it secures and supports long-term regulatory confidence.
Looking ahead: AI-powered chemistry in 2030
As these systems converge, AI-powered chemistry will manifest as highly integrated workflows where software schedules an experiment, captures analytics, and proposes the next conditions with quantified uncertainty – while chemists remain in the driver’s seat to set objectives, constraints, and sanity-check the path. As Dai et al. confirmed, modular autonomous platforms already show that this is feasible in practice. This active-learning autonomy is explicitly framed to close the gap between computational screening and experimental realization.